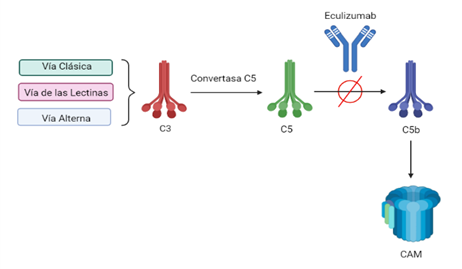
El eculizumab fue pionero en el tratamiento de la hemoglobinuria
paroxística nocturna (HPN); sin embargo, con el paso de los años
se ha descubierto que este medicamento también ha sido útil en
el tratamiento de otras patologías como el síndrome urémico
hemolítico atípico (SUHa), la glomerulonefritis C3, la
preeclampsia con el síndrome HELLP, la miastenia gravis
generalizada refractaria, el trastorno del espectro de la
neuromielitis óptica (TENMO) y también la anemia hemolítica por
crioaglutininas.
Algunas de las enfermedades más relevantes y con mayor cantidad
de estudios asociados al uso del eculizumab se describen en
detalle a continuación.
Hemoglobinuria Paroxística Nocturna (HPN). Es probablemente la
enfermedad que más se asocia al uso terapéutico del eculizumab y
entre sus manifestaciones clínicas se encuentra la anemia
hemolítica, fallo en la médula ósea y trombofilia23. Se da por una
mutación adquirida en un gen del brazo corto del cromosoma X, el
cual codifica para glucosil-fosfatidil-inositol (GPI), a partir
de la expansión clonal de las células madre mutadas de un clon
hematopoyético con la deficiencia en proteínas que normalmente
están unidas a la membrana por el GPI24. Las células provenientes de
esta célula madre pueden tener una deficiencia total (tipo III)
o parcial (tipo II)25.
El GPI se encuentra en la membrana y algunos de los factores que
se unen a esta molécula son el CD55 y el CD59, encargados de
regular el sistema del complemento. Los pacientes con HPN no
presentan estos factores, por lo que la actividad del
complemento no puede detenerse y en consecuencia presentan
hemólisis26.
La destrucción de los eritrocitos hace que se produzca anemia y
que el paciente presente síntomas como fatiga, dolor abdominal,
disfagia, disfunción eréctil, disnea, hemoglobinuria y
trombosis. El único tratamiento conocido que resulta en la
completa recuperación del paciente es el trasplante de médula
ósea, pero para este procedimiento se necesita un donante
compatible y la supervivencia es de un 50% a 60%2, por esta razón, este
tipo de tratamiento se limita normalmente a pacientes jóvenes y
con donadores muy específicos26.
Alrededor de un 50% de las personas diagnosticadas con esta
enfermedad mueren26,
la trombosis es uno de los mayores riesgos, particularmente en
venas cerebrales y hepáticas. La expectativa de vida, una vez
diagnosticada es aproximadamente 10 años contrario a 8 años en
personas no tratadas24. Muchos pacientes son
dependientes de transfusiones, porque es uno de los principales
abordajes que se realizan; sin embargo, este tipo de tratamiento
puede resultar contraproducente por los riesgos que
representa26.
El tratamiento con eculizumab disminuye o puede llegar a eliminar
la necesidad de transfusiones de sangre3 hasta en un 74%27; además reduce el
riesgo de trombosis. Hasta el momento, este fármaco es el único
aprobado por el FDA para el tratamiento de HPN3; sin embargo, hay
estudios de otros medicamentos que se pueden estar desarrollando
con el mismo fin. A pesar del uso del fármaco, la anemia y algo
de destrucción extravascular va a seguir ocurriendo, porque no
son blancos del medicamento y están asociadas a otras
deficiencias26.
Como se mencionó, la hemólisis extravascular en estos pacientes
se presenta por el depósito de C3d en los glóbulos rojos y
podría ser lo que explica que los pacientes tratados con
eculizumab obtengan un examen antiglobulina positivo directo,
pero uno de IgG negativo. Además, desarrollan una anemia
moderada y un recuento de reticulocitos alto28.
Por esta razón, es importante que los pacientes que tomen el
medicamento sean monitoreados con un recuento de reticulocitos,
lactato deshidrogenasa (LDH), recuento completo de glóbulos
rojos y un perfil bioquímico semanalmente durante las primeras
cuatro semanas y luego una vez al mes28. Esto se hace de esta manera
porque esos son los tiempos en que se administran las dosis.
Se recomienda que el paciente sea tratado con 600 mg de
medicamento en las primeras cuatro semanas y luego, se continúe
administrando cada dos semanas una dosis de 900 mg. El suero
concentrado del medicamento debe ser de 35 μg/mL18.
Tras la administración del fármaco, se observa una mejoría casi
inmediata en los pacientes29, con una disminución
significativa de los niveles de LDH (indicativo de la hemólisis
intravascular), además se reducen o eliminan las transfusiones
de sangre, se estabilizan los niveles de hemoglobina y mejora la
fatiga. Al cabo de dos semanas se pueden alcanzar niveles casi
normales en la mayoría de las variables25.
En resumen, los únicos dos métodos para curar esta enfermedad son
el trasplante de médula ósea alogénico y el uso de eculizumab
permanentemente30,
por eso, contar con este medicamento es una opción menos
invasiva para mejorar la calidad de vida de los pacientes, sin
generar efectos secundarios significativos23.
En cuanto a la seguridad y eficacia de HPN se han hecho estudios
para determinar la tolerabilidad y toxicidad del eculizumab,
dentro de los cuales se han observado los siguientes efectos
adversos: cefalea, nasofaringitis, náuseas, pirexia, mialgia,
fatiga, herpes simple2, infección urinaria, infección
respiratoria, sinusitis, infección vírica, trastornos
psiquiátricos, insomnio, trastornos del sistema nervioso,
mareos, dolor faringolaríngeo, tos, trastornos
gastrointestinales, prurito, trastornos osteomusculares y del
tejido conjuntivo, dolor de espalda, artralgia, dolor de las
extremidades y calambres musculares22. La mayoría de los efectos se
presentan como leves o moderados2,4, siendo la
septicemia meningocócica la más grave2, e incluso, puede inducir a la
muerte.
Síndrome Urémico Hemolítico Atípico (SUHa). Esta es otra
enfermedad para la cual el eculizumab fue aprobado. El SUHa se
clasifica dentro de las microangiopatías trombóticas (TMA) y su
detección es bastante difícil, pues sus síntomas pueden
traslaparse con otros de enfermedades relacionadas con esta
categoría. Los síntomas incluyen: anemia hemolítica,
trombocitopenia no inmune y fallo en órganos. Se da
mayoritariamente en niños entre los seis meses y los cinco años,
pero también puede darse en adultos, mayoritariamente en
mujeres3.
En sí, lo que ocurre con esta enfermedad es una hiperactivación
de la vía alterna del complemento (en la C3 convertasa) y una
pérdida en la función de regulación. El C3 se sigue hidrolizando
a C3b y esto conlleva a una deposición de C3b en la CAM, lo que
produce eventualmente daño tisular31.
Como se puede observar, el síndrome es causado por defectos en la
regulación del sistema del complemento. Estos defectos pueden
ser adquiridos, heredados o una mezcla de ambos y lo que
producen es la activación crónica e incontrolada del sistema del
complemento, por lo cual se desencadena la activación de
plaquetas, leucocitos y células endoteliales, además de
microangiopatía trombótica sistémica, que afecta a múltiples
órganos como el sistema nervioso central, riñones, corazón y
tracto gastrointestinal (TGI)32.
Antes de que se descubriera que el eculizumab podía tratar el
SUHa, el rango de mortalidad era muy alto (33% a 40%) durante
las primeras manifestaciones clínicas32. Los pacientes llegaban hasta
la etapa final de enfermedad renal y el tratamiento incluía
infusiones de plasma33, que, si bien ayudaban a
mantener los niveles de plaquetas y LDH, la desregulación del
complemento y la microangiopatía trombótica se mantenían,
haciendo que la cura no fuera del todo exitosa32. Además, después
del primer año de tratamiento con plasma, se generaba daño renal
permanente, una progresión en la enfermedad renal terminal o
muerte32.
Una vez que los pacientes inician el tratamiento con eculizumab,
se puede observar que los niveles de hemólisis disminuyen
(medidos por LDH), el recuento de plaquetas aumenta y la
creatinina disminuye y después de una semana, es posible que el
paciente deje la terapia con plasma. Esto se termina de
confirmar cuando después de 20 días, los niveles de hemólisis y
plaquetas están normales y la creatinina se acerca mucho a la
normalidad33.
La remisión clínica se da después de 60 días de tratamiento con
el fármaco33. Se
recomienda que la concentración de suero sea de 50 a 100
µg/mL25, además de
una dosis de 900 mg semanalmente durante el primer mes y luego,
a la quinta semana, la dosis debe ser de 1200 mg y continúa cada
dos semanas6. Cabe
señalar que la dosis de este medicamento es más alta para los
pacientes con SUHa que para los pacientes con HPN.
Las pruebas que se han realizado, demuestran que la inhibición
del complemento terminal mejora la función renal, aun en
aquellos pacientes donde su función renal estaba seriamente
comprometida por la transferencia de plasma. Entre más rápido se
intervenga al paciente con el eculizumab, mayores beneficios
podrá tener y revertir el daño en órganos32.
También se demostró que el uso de eculizumab aumenta la tasa de
filtración glomerular (TFG) estimada. Por esta razón, los
pacientes eran eximidos de la diálisis, lo que significó un
notable aumento en su calidad de vida. Es válido agregar que no
se han observado acumulaciones de desechos tóxicos ni
infecciones relacionadas con las aplicaciones de este
medicamento, lo cual revela que su uso es seguro32.
La tolerabilidad del eculizumab es satisfactoria y se han
reportado pocos casos de efectos adversos graves relacionados
con el tratamiento. Algunos de estos pocos efectos son la
hipertensión maligna, hipertensión severa, bacteriuria
asintomática, infección por influenza, peritonitis, esclerosis
venosa en el punto de infusión y fiebre34.
El SUHa es una de las enfermedades más tratadas con eculizumab;
razón por la cual se han hecho pruebas clínicas suficientes que
comprueban su seguridad a largo plazo. Estos estudios se
realizaron tanto en pacientes pediátricos como con adultos
tratados con eculizumab durante los primeros cinco años del
registro hasta enero del 201735.
El grupo control (pacientes no tratados con eculizumab)
presentaron infecciones severas a diferencia de los que
recibieron la droga, esto en pacientes pediátricos; sin embargo,
no hubo nuevos descubrimientos en cuanto seguridad y se probó un
riesgo-beneficio positivo35.
Otros estudios afirman que el uso del eculizumab por un período
corto es más seguro y efectivo y concluyen que extender las
dosis del medicamento puede exceder la dosis terapéutica en
sangre36. También se
hicieron estudios aplicados exclusivamente a pacientes
pediátricos, los cuales arrojaron resultados excelentes, pues no
había tanta aparición de efectos secundarios37.
Miastenia gravis generalizada refractaria. Otra de las
enfermedades en las que Soliris® ha resultado efectivo. La
enfermedad generalizada es una transmisión sináptica
neuromuscular autoinmune crónica mediada por un anticuerpo y su
incidencia, al igual que las otras enfermedades, es baja.
Esta patología se caracteriza por una debilidad en los músculos y
fatiga que son causadas porque los anticuerpos están dirigidos a
los receptores del músculo esquelético y las proteínas de las
uniones neuromusculares. Otros síntomas son la dificultad para
hablar, tragar o masticar, falta de aire, caída de uno o ambos
párpados y visión borrosa3.
A nivel inmune, las personas que padecen de esta enfermedad
tienen anticuerpos contra el receptor de acetilcolina (AChR),
los cuales se pueden unir a factores que inducen la CAM, lo cual
genera el daño en las membranas de los músculos y afecta la
neurotransmisión3.
Los tratamientos que se usan normalmente son los inhibidores de
acetilcolinesterasa, como transfusiones de plasma o
inmunoglobulinas intravenosas, terapia inmunosupresora (con
corticosteroides, ciclosporina, entre otros) y timectomía. Sin
embargo, estos tratamientos no mejoran la calidad de vida de
estos pacientes de forma significativa, en especial para las
personas con alguna resistencia al tratamiento que requieren
terapias más agresivas para las crisis que pueden resultar en su
muerte3.
Como se puede observar, no se contaba con un tratamiento efectivo
hasta que se utilizó el eculizumab. La dosis recomendada es de
900 mg cada semana por cuatro semanas en la fase inicial y luego
1200 mg cada dos semanas hasta que el médico lo indique en la
fase de mantenimiento3.
Con respecto a la seguridad y eficacia del medicamento para esta
enfermedad, los pacientes evaluados no presentaron adversidades
ni registros de infecciones; por el contrario, se observó una
notoria reducción en la sintomatología, lo cual reafirma la
eficacia del medicamento38.
Trastorno del Espectro de la Neuromielitis Óptica (TENMO). Esta
es una enfermedad autoinmune que afecta el sistema nervioso
central (desmielinizándolo) y puede llegar a atentar contra la
vida del paciente. Se descubrió que la enfermedad es producto de
un auto anticuerpo IgG autorreactivo específico que es dirigido
contra el canal de agua de la acuaporina 4 (AQP4) en el suero de
unas 75% a 90% de los pacientes39.
Los pacientes con TENMO sufren ataques de neuritis y/o mielitis
óptica que puede llegar a incapacitar al paciente y aunque
existe cura, el problema radica en que no todas las personas
llegan a experimentar una recuperación completa pues tienen la
posibilidad de recaídas39.
Algunos estudios aseguran que esta enfermedad se presenta en
personas de cualquier edad, pero es más común a partir de los 40
años. Las personas no caucásicas son las más afectadas y las
recaídas se dan con mayor frecuencia en pacientes femeninas39.
A lo largo de los años, esta enfermedad ha ido evolucionando en
su diagnóstico y detección. Anteriormente se creía que era una
variación de la esclerosis múltiple, ahora es posible
reconocerla como una entidad clínica separada, lo cual ha
permitido que su tratamiento y detección sean más fáciles y
favorables para los pacientes que la sufren, pues como en todas
las enfermedades, la detección temprana es algo vital39.
El eculizumab ha sido el primer tratamiento aprobado para tratar
esta enfermedad. Es importante agregar que el uso de este
medicamento ha disminuido el tiempo en los hospitales y las
recaídas; por tanto, ha contribuido en forma significativa a
mejorar la calidad de vida de los pacientes39.
Síndrome de Guillain Barré (SGB). Es una polineuropatía
inflamatoria autoinmune que suele ser antecedida por una
infección viral, bacteriana e incluso se ha relacionado con
algunas inmunizaciones. Entre los agentes infecciosos más
comunes están el virus del Chikungunya, la influenza, Virus de
Inmunodeficiencia Humana (VIH), poliovirus circulante tipo 3,
citomegalovirus, el virus de Epstein Barr, recientemente el
SARS-CoV-2 y bacterias como la Mycoplasma pneumoniae y la
Campylobacter jejuni40,41. Los estudios
electrofisiológicos permiten distinguir varios subtipos; entre
los más comunes se encuentran: la polirradiculoneuropatía
desmielinizante inflamatoria aguda (PDIA), la neuropatía axonal
motora aguda (NAMA) y la neuropatía axonal sensitivo-motora
aguda (NASMA)42.
Los síntomas del SGB pueden empeorar rápida y abruptamente,
porque, así como pueden presentarse en tres o cuatro semanas,
también pueden desarrollarse en cuestión de horas o días.
Comúnmente la debilidad y parálisis muscular inicia en piernas y
se disemina a los brazos (parálisis ascendente)40.
Generalmente, los primeros síntomas son entumecimiento y
debilidad muscular, después puede haber pérdida de reflejos,
movimientos descoordinados y en los casos más graves, el
paciente puede presentar parálisis total, problemas
respiratorios y cardíacos. Afortunadamente, la mayoría de
pacientes logra recuperarse de casos graves por medio de
fisioterapia y tratamiento que consta principalmente de
inmunoglobulinas intravenosas y plasmaféresis40,43.
El mecanismo inmunológico por el cual se activa el SGB es el
mimetismo molecular que existe entre los gangliósidos presentes
en los nervios periféricos y los antígenos microbianos. Los
gangliósidos son glicoesfingolípidos con residuos de ácidos
siálico, que se encuentran en la superficie de la membrana
aportando protección y estabilidad al axón-mielina y actúan
también como receptores funcionales de proteínas presentes en
las vainas de mielina40,44.
Ahora bien, ese parecido estructural entre los gangliósidos y los
antígenos microbianos produce una respuesta inmune cruzada. Los
anticuerpos antes producidos para atacar la infección actúan
ahora contra los gangliósidos presentes en los nervios
periféricos, activando el complemento y provocando la
degeneración axonal en los nodos de Ranvier, en el caso de la
variante NAMA y la destrucción de las vainas de mielina, en la
variante PDIA, en donde las células Schwann son el antígeno
blanco para estos auto anticuerpos42,45.
Dado que la respuesta inmune cruzada en el SGB conlleva a la
activación del complemento y por tanto a la activación del CAM,
que es justamente lo que inhibe el eculizumab, es recomendable
realizar más estudios con el objetivo de investigar si la
adición del eculizumab al tratamiento convencional para
pacientes con SGB, disminuye sus síntomas, tal y como se
describe en el artículo “Safety and eficacia of eculizumab in
Guillain-Barré syndrome: a multicentre, double-blind, randomised
phase 2 trial” estudio realizado en 13 hospitales de Japón45.
Síndrome Antifosfolípido Catastrófico (CAPS). También denominado
como “Síndrome de Asherson”46, es una enfermedad autoinmune
rara y poco común que se ha descrito como una manifestación
grave del síndrome antifosfolípido (SAF) que cursa con falla
multiorgánica47
secundaria a la aparición de trombosis microvascular en
distintos órganos, en menos de una semana48.
La enfermedad se presenta en menos del 1% de los casos de SAF46,49. Esta afección es una situación
médica urgente que requiere de seguimiento y tratamiento
temprano46. El CAPS
se manifiesta en un 70% de los casos en mujeres, también se ha
observado en pacientes de edad avanzada y recién nacidos47,50.
La patogenia del CAPS no se comprende por completo46,50, pero dentro de las teorías
propuestas se encuentra la del mimetismo molecular, que ocurre
después de la infección con microorganismos que presentan
similitud estructural de sus péptidos con los del hospedero. A
nivel inmunológico, en las personas que padecen esta enfermedad,
los anticuerpos antifosfolípidos (aPL) se unen a las superficies
de los fosfolípidos por medio de proteínas intermediarias de
unión a lípidos50,
como anti-β2-glicoproteína I (GP1)46. El complejo proceso implica la
activación del receptor 4 tipo Toll (TLR-4), que induce una
tormenta de citoquinas46, seguido por la activación de
células endoteliales, plaquetas, monocitos, células
trofoblásticas y neutrófilos con implicaciones
protrombóticas49.
Se ha demostrado que los aPL tienen la capacidad de inducir un
estado de hipercoagulabilidad por medio de distintos mecanismos
de acción, dentro de los que se encuentra la activación del
complemento, el cual, a su vez, juega un papel importante en la
patogenia de varias microangiopatías trombóticas51.
Según los expertos, los pacientes con esta afección deben ser
tratados con anticoagulantes y corticosteroides, para lo cual se
considera el uso adicional de inmunoglobulina intravenosa (IVIG)
o intercambio de plasma (plasmaféresis). No obstante, existen
informes de casos en pacientes con CAPS persistente y que, a
pesar del tratamiento triple, no presentan mejoras49.
Por su parte, el empleo del eculizumab como inhibidor del
complemento para la intervención del CAPS, ha mostrado prever
eficientemente la progresión hacia la tormenta micro
trombótica51. No
obstante, es importante evaluar sus posibles efectos
secundarios52.